Химия. Химия органическая
X. Химия органическая. В настоящее время под химией органической разумеют отдел химии, занимающийся изучением соединений углерода, как входящих в состав организмов — растительных и животных, так и получаемых искусственно. Из этого определения следует, что органические соединения подчиняются всем общим законам химии; лишь закон кратных отношений, благодаря большой сложности органических соединений, утрачивает применительно к ним свой простой и ясный смысл. Выделение химии органической в особую науку делается из соображений удобства и вызывается громадным числом органических соединений (около 200 000), их своеобразным характером, проявляющимся особенно в том, что среди них очень распространена изомерия, т. е. различие свойств при одинаковом составе и молекулярном весе, и громадным значением этих соединений как для биологии и техники, так и для развития теоретических воззрений на природу сродства и строение молекул. К этому следует добавить, что своеобразие органических соединений, вызываемое присутствием в них углерода, обусловливает необходимость особой методики при работах в этой области. Но к такому взгляду на органические соединения пришли долгим путем и лишь в XIX столетии, хотя с ними человечество познакомилось очень давно.
Наши сведения о знакомстве древних народов с органическими соединениями отрывочны и неполны. Но все же известно, что древние знакомы были с добыванием тростникового сахара из сахарного тростника, употреблением растительных масел для освещения и смазывания тела; с камфарой, терпенами, смолами и бальзамами и употреблением их при религиозных церемониях и для бальзамирования трупов, с пахучими веществами, с различными экстрактами растений — ядовитыми и целебными; с красками из растений (ализарин, индиго) и животных (финикийский пурпур, кармин), с кислотами уксуса и прокисшего молока. Знали также и некоторые технические процессы переработки органических веществ: приготовление вина и пива брожением, мыла из жиров и пр. Но почти все эти вещества были известны в виде смесей. Кроме того, до XVII ст. органические соединения не отличали от неорганических. Но мало-помалу установлены были существенные отличия минеральных веществ от веществ растительных и животных. Главнейшие, бросавшиеся в глаза, отличия сводились к способности последних гореть, подвергаться процессам брожения, гниения и тления и к поведению их при сухой перегонке. Так как в то время понятие об элементах в современном смысле еще почти отсутствовало, различия эти объяснялись преобладанием в органических соединениях начал (элементов в духе Аристотеля) водного и горючего, а в минеральных землистого.
Но вот, на пороге XIX века, Лавуазье реформирует химию. Утвердив и расширив введенное Бойлем понятие об элементе в современном смысле и установив состав воды и угольного ангидрида, Лавуазье положил начало анализу органических соединений и показал, что в громадное большинство веществ растительного происхождения входят только углерод, водород и кислород; в веществах животного происхождения к этим трем элементам присоединяется азот. Из этих четырех элементов состоит большинство природных органических соединений. Их и называют органогенами. С методологической стороны органический анализ становится одной из основ дальнейшего развития химии органической. Ряд ученых способствует развитию методов органического анализа, и в 40-х годах XIX века Либих придает ему почти окончательную форму. Итак, объяснять различия органических соединений от минеральных различием элементов оказалось невозможным.
Пытались объяснять это различие различием законов, имеющих место при образовании минеральных и органических соединений. Говорили, что простые количественные законы постоянства состава и кратных отношений неприменимы к органическим соединениям. Но успехи органического анализа заставили отказаться и от этого объяснения.
Оставалась еще одна идея, которая упорно господствовала и опираясь на которую можно было проводить глубокую, принципиальную грань между органической и неорганической химией — это идея жизненной силы (см. витализм). Допускалось, что органические соединения образуются под влиянием особой, организмам присущей жизненной силы. Вера в жизненную силу поддерживалась особым направлением изучения органических соединений в первой половине XIX века. Тогда как минеральные соединения можно было получать из более простых и из элементов, органические можно было изучать только в одном направлении, именно, подвергая упрощению сложные, добытые из организмов вещества. Фраза: «природа созидает, химик разрушает» (Жерар), хорошо выражает мнение химиков первой половины XIX века по этому вопросу. Первым классическим примером синтеза вещества, ранее добывавшегося из организмов, является синтез мочевины, осуществленный Велером в 1828 г. В настоящее время мочевина, представляющая прекрасное удобрение, готовится синтетически в техническом масштабе. Знаменитый синтез Велера был первым шагом на пути синтеза органических веществ. Правда, его одного было недостаточно, чтобы разрушить веру в жизненную силу и подвести превращения органических соединений под общие законы химических превращений; но по этому пути пошли другие. Особенно важных общих результатов достиг М. Бертло, который разработал в половине XIX века общие приемы синтеза различных классов органических веществ из элементов или из простейших соединений — углекислоты, воды, аммиака. Этот путь привел к синтезу важнейших природных веществ различных классов и еще большего числа соединений, не найденных в природе. Углеводороды, спирты, кислоты, сахара (Э. Фишер); жиры (Бертло), алкалоиды, яркие краски: ализарин, индиго; нежнейшие ароматы растений, искусственный каучук — получены синтетически, и некоторые приготовляются технически в таком широком размере и столь высокого качества, что вытесняют продукты, добываемые из организмов. Очередь за синтезом немногих алкалоидов (хинина), сложных углеводов и, особенно, белковых веществ и неорганизованных ферментов. Но нет поводов сомневаться, что в ближайшем будущем и эти последние задачи будут разрешены; для устранения же надобности прибегать к жизненной силе — сделанного до сих пор более, чем достаточно. При образовании органических веществ всюду действуют те же силы и по тем же законам, как и при образовании соединений всех остальных элементов.
Теоретические основы химии органической развились за очень короткий период первых трех четвертей XIX века. Но в это время развитие шло бурным путем, различные теории быстро сменяли одна другую, каждая внося известную долю истины в последующую. При этом в самом начале химия органическая развивалась медленнее неорганической и под ее влиянием, но чем далее шло ее развитие, тем более ускорялся темп его и углублялись ее воззрения. Для неорганической химии первой половины XIX века особенно характерен дуалистический характер. Лавуазье на первое место поставил кислород и кислородные соединения. Элемент, соединенный с кислородом, получил название радикала (Base). Показав, что органические кислоты состоят из углерода, водорода и кислорода, Лавуазье рассматривал их как аналоги минеральных кислот (собственно ангидридов) с тем отличием, что в минеральных кислотах радикал простой, а в органических — сложный. Этим положено было начало теории сложных радикалов. Теория эта разделялась многими химиками, и еще в 1837 году знаменитые Дюма и Либих так выражали свой взгляд на задачи химии органической: «Как мы ее понимаем, химия органическая представляет нам радикалы, играющие ту же роль, как металлы, и другие, играющие роль, сходную с ролью кислорода, хлора, серы. Эти радикалы, соединяясь между собой или с элементами, порождают при помощи самых простых законов минеральной химии все органические соединения». В открытии этих радикалов и их изучении видят они важнейшую цель химии органической. Как главной задачей неогранической химии являлось разложение сложных веществ (Scheidekunst) и открытие новых элементов, так в химии органической — открытие радикалов.
Открытие Гей-Люссаком радикала циана и получение свободного циана, классическая работа Либиха и Велера над соединениями бензоила (С6Н5СО'), (черточки указывают свободную валентность, а не заряд, т. к. радикалы в отличие от сложных, ионов электрически нейтральны), Бунзена над какодилом (CH3)2AS', попытки получения этила и пр. относятся к этой области. Понятие о радикале не утратило своего значения и до сих пор, и даже с открытием в 1900 году Гомбергом трифенил метила (С6Н5)3С' — настоящего свободного радикала — и затем ряда других соединений той же группы, а также азотсодержащих свободных радикалов, например (С6Н5)2N', (СН3)4N’, Виландом и другими — интерес к радикалам весьма увеличился. Исследования этого рода имеют большое значение в связи с коренными проблемами о валентности элементов, о сложном строении атомов и разложимости элементов. Но для развития химии органической теория сложных радикалов, носившая дуалистический характер, и само дуалистическое воззрение, наиболее ярко выражавшееся в электрохимической теории Берцелиуса, оказались недостаточно широкими. Для теории сложных радикалов весьма важным являлся вопрос об электрохимическом характере элемента и радикала, и она принимала, что элементы противоположного электрохимического характера должны играть в соединении существенно различную роль.
Новые и многочисленные факты, касающиеся способности электроотрицательных элементов, особенно хлора, замещать в органических соединениях водород без изменения существенных свойств, т. е. явление, изученное особенно Дюма и Лораном и получившее название металепсии, явилось резким противоречием дуалистическому воззрению на органические соединения, заставив признать, что электроотрицательный элемент хлор может заменять электроположительный водород, становясь на его место, и играть его роль. Это мнение, встретившее сперва противодействие, укрепилось и вызвало появление особых взглядов на органические соединения, которые выразились как в недолговечной теории ядер Лорана, так и в первоначальной теории типов Дюма. По Лорану, органические соединения заключают в себе ядра из атомов, из которых простейшие состоят только из углерода и водорода. Ядра эти можно представить в виде призм, вершины которых заняты атомами углерода, а ребра атомами водорода. Из этих простейших ядер могут происходить их производные замещением атомов водорода атомами других элементов или сложными группами. Так как основная форма при этом сохраняется; то производные ядра сходны с первоначальными. Кроме изменения путем замещения, ядра могут изменяться и другим путем, присоединением атомов элементов и сложных групп, что можно изобразить геометрически как насаждение пирамид на стороны призм. Глубокая идея, лежащая в основании этого воззрения, что свойства органического соединения обусловливаются не столько сходством элементов, сколько сходством распределения атомов, не нашла в свое время отклика. Ту же почти мысль высказывает Дюма в первоначальной теории типов. Он уподобляет органические соединения планетным системам, в которых отдельные атомы играют роль планет, удерживаемых вместе силой взаимного притяжения. Если какой-либо атом замещается атомом другого элемента или даже сложной группой, система остается неизменной, и все соединения, получающиеся таким путем, относятся к одному типу. Теории эти развивались в половине тридцатых годов XIX века.
Дальнейшие существеннейшие шаги в развитии химии органической заключались в разграничении понятий об эквиваленте и атоме, в проведении понятия о частице и в выражении частиц органических соединений формулами, отвечающими в парообразном состоянии двум объемам, чем особенно химия обязана Лорану и Жерару.
Выдвинув на первый план понятие о химической частице, они придали первенствующее значение изучению химических реакций, особенно же реакций замещения или двойных разложений. Придав особый смысл явлению замещения атомов сложными группами, они ввели в теорию типов и замещения радикалы, придав им новый смысл, не требовавший способности их к существованию в свободном состоянии, и присвоив им особое название «остатков». Раз изучено было много химических превращений и выражено было уравнениями, отвечающими частичным количествам тел, — между превращениями разных веществ стали обнаруживаться черты сходства, ранее не замечавшиеся. Между этими чертами сходства разных групп превращений особенно важными для дальнейшего развития теории оказались те, которые сближали превращения органических соединений с превращениями простейших минеральных соединений: водорода, соляной кислоты, воды и аммиака. Сложные вещества оказалось возможным рассматривать как происшедшие из этих типов замещением водорода их сложными группами. Теория типов обязана своим возникновением и развитием Жерару, в ее рамки хорошо укладывались известные в 50-х годах прошлого столетия органические соединения. Она не только давала возможность сближать сходные между собой соединения, относя их к одному типу, но позволяла понимать различие таких соединений, которые, имея одинаковый качественный и количественный состав и одинаковый частичный вес, являются различными. Такое различие понималось или как следствие принадлежности этих соединений к разным типам, или, при одинаковости типа, как следствие различия радикалов. Различие соединений при одинаковости качественного и количественного состава есть явление, давно подмеченное среди органических соединений и чрезвычайно распространенное: оно получило название изомерии в широком смысле слова. Более подробное изучение привело к отличию полимерии, когда частичные веса при одинаковом составе различны, и изомерии, когда они одинаковы.
Теория строения органических соединений, по выражению одного выдающегося ученого, принадлежит к возвышеннейшим произведениям человеческого разума. Теория эта сделала химию органическую сродной математике, хотя в то же время она не утратила близости и с искусством, т. к. дает обширное поле воображению. Теория строения опирается на понятие о валентности или атомности элементов. Понятие это развилось постепенно, и в этом отношении важную роль сыграли работы над многоатомными радикалами, исследования Франкленда над металлоорганическими соединениями, показавшие, что способность атомов соединяться с определенным числом атомов другого элемента есть их характерное свойство, и, наконец, воззрения Кольбе, который производил органические соединения от углекислоты.
Основания теории строения, как это часто бывает с научными идеями, почва для которых подготовлена и которые, так сказать, висят в воздухе, были высказаны одновременно знаменитыми учеными разных стран: в Англии - Купером, в Германии — А. Кекуле и у нас — А. М. Бутлеровым, который особо много сделал для ее развития и распространения, как лично, смело, строго проводя ее, идя впереди Кекуле, так и создав школу русских химиков, посвятивших себя ее дальнейшей разработке. Сущность теории строения, по выражению Кекуле, сводится к признанию, что «в частицах отдельные атомы связаны не со всеми или не один из них со всеми, а каждый из них соединен с немногими соседними атомами, как в цепи звено со звеном».
Первым положением теории строения является признание четырехатомности углерода, вторым — способности углеродных атомов соединяться друг с другом, затрачивая на это большее или меньшее число единиц атомности. Формулы, которые, опираясь на эти положения, выражают взаимные отношения атомов, образующих молекулы, называются структурными, конституционными или рациональными. Для определения строения соединений с одним атомом углерода достаточно первого положения и знания атомности соединенных с углеродом элементов. Как примеры, приведем следующие соединения:
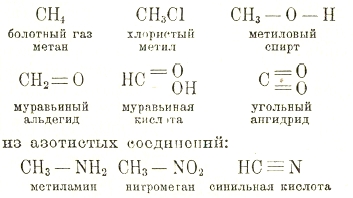
Все эти соединения суть насыщенные по отношению к углероду, все единицы сродства углеродного атома затрачиваются на связь с другими. Углеводород СН4 есть углеводород предельный, неспособный к дальнейшему соединению.
В соединениях с несколькими атомами углерода принимают цепи из атомов углерода:
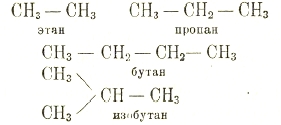
Все эти соединения углерода с водородом суть углеводороды предельные, неспособные к присоединению.
Состав всех их можно выразить формулой СnН2n+2. Склонность к соединению атомов углерода друг с другом весьма велика — получен углеводород С66Н122. Представив себе, что водород в этих углеводородах замещен элементами или сложными группами-радикалами 'ОН, NH2'. NO2' и т. д., получим множество разнообразных соединений, являющихся производными предельных углеводородов:
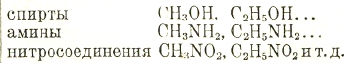
Все эти соединения суть предельные или соединения жирного ряда. На этом основании химию органическую определяют иногда как химию углеводородов и их производных. Кроме предельных соединений, известно множество соединений более бедных водородом, например С2Н4 этилен, С2Н2 ацетилен. Это — представители ближайших к предельным рядов углеводородов; существует много углеводородов и еще более бедных водородом. Этилен и ацетилен весьма склонны вступать в реакции прямого соединения с водородом, галоидами, окислами азота, с элементами воды и перекиси водорода и пр. Известно множество более сложных углеводородов с подобными свойствами; в них этиленовая и ацетиленовая группировки могут входить в частицу несколько раз.
Вопрос о характере связи в этилене и ацетилене, обусловливающем их особенный характер, представляет большие трудности, чем о связи в предельных соединениях, и решается различно. В предельных соединениях каждый атом углерода затрачивает на связь с соседним по одной из своих четырех валентностей: такая связь называется простой (или одиночной) и характеризуется прочностью. В этилене приходится признать или присутствие свободных, ненасыщенных валентностей, по одной у каждого атома углерода, или двойную связь между атомами углерода (но не равную двум простым), или, наконец, признать трехвалентиый углерод. Соответственно, формулы этилена будут:
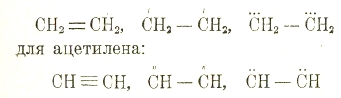
Обычно изображают этиленную и ацетиленную связь первым способом, но имеют ввиду ее особенности, отчасти объясняемые стереохимией и новейшими теориями валентности. От углеводородов с двойными и тройными связями, как и от предельных, известно много производных.
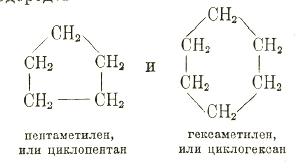
Кроме образования прямых (нормальных) или разветвленных (изо) цепей, атомы углерода могут быть связаны кольцеобразно. Наибольшей прочностью обладают кольца из 5 и 6 атомов углерода. Кольца из 5 и 6 атомов углерода распространены в природе (кавказской нефти, растениях и пр.) и играют большую роль в технике (лекарства, краски, взрывчатые вещества). Особенно важны производные углеводорода бензола С6H6 (см.), получившие название ароматических соединений (см.).
Теория строения достигла своего кульминационного пункта, когда А. Кекуле, выяснив циклическое строение бензола, создал теорию ароматических соединений, которая изящно разрешила сложный вопрос об отношениях и реакциях веществ этой группы и сделалась путеводной звездой при работах в этой области; сюда направлен был главный поток сил химиков в последние 30 лет прошлого столетия, особенно в Германии.
Кекуле придал бензолу такую формулу:
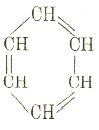
Эта формула, по существу, и до сих пор является общепринятой. Особенно важным в ней является циклическое расположение шести равнозначных групп СН, вопрос же о том, как используются остальные валентности атомов углерода, имеет меньшее значение. Другими учеными предлагались и несколько иные формулы строения:
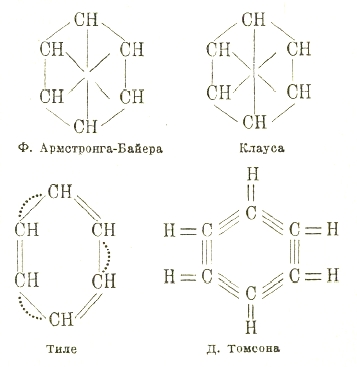
В последней формуле предполагается, что простой связи отвечают пара общих электронов, поэтому каждой черте в других формулах отвечают две черты в последней формуле.
Все формулы выражают, по существу, то же, что можно выразить такой формулой:
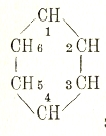
Эта простая формула совершенно достаточна для объяснения изомерии среди ароматических соединений.
Исходя из строения бензола, оказалось возможным выразить строение других углеводородов, обладающих ароматическим характером. Таких углеводородов известно очень много, и многие из них имеют большое значение в технике, например: трифенил-метан, нафталин, антрацен и др. Позднее были получены и изучены более простые (с меньшим циклом) и более сложные углеводороды циклического строения — содержащие более водорода, например, триметилен и другие полиметиленовые углеводороды
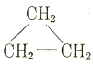
и углеводороды с двойной связью в цикле, например
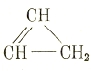
Комбинации различных циклических систем встречаются в сложных природных углеводородах — терпенах С10Н16 и сексвитерпенах — С15Н24, а производные углеводородов
составляют главную массу кавказской нефти. Далее оказалось, что циклические соединения могут содержать в цикле не только углерод. Такие циклы получили название гетероциклов. Азотистые гетероциклы очень распространены в живой природе, входят в состав белков, хлорофилла растений, красящего вещества крови — гемоглобина и алкалоидов растений. К простейшим азотистым гетероциклам принадлежат пиррол

пиридин и продукты присоединения к ним водорода — пирролидин
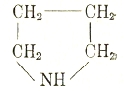
и
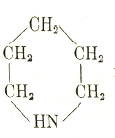
пиперидин.
Теория строения сыграла и продолжает играть в химии органической громадную роль. Пользуясь ею, не только удается выяснить причину изомерии громадного числа соединений и установить на общих началах их соотношения, но и сознательно выбирать пути к синтезу новых органических соединений, ибо из громадного числа мыслимых сочетаний атомов способными к существованию оказываются те, которые удовлетворяют основным принципам теории. Теория строения, изображая структурными формулами способ связи между атомами, не касалась вопроса о расположении их в пространстве.
Установление строения соединений сделалось одной из важнейших и труднейших задач химии органической. Для решения подобных вопросов в отдельных случаях потребовалось много десятков лет работы выдающихся исследователей. Примерами подобного рода могут служить терпены — С10Н16, камфара С10Н16О, хинин С20Н24N2О2 и пр. Задачи подобного, рода тем труднее, что тут не имеется общих шаблонных методов. Руководствуются при этом как физическими свойствами, так и разнообразнейшими реакциями. При этом молчаливо допускают, что превращения захватывают обычно определенную ограниченную часть молекулы, а остальное распределение связей сохраняется. Это действительно обычно имеет место в силу прочности углерод-углеродной связи, но далеко не всегда. Иногда решение вопроса затрудняется особой тонкой изомерией-таутомерией (см.), когда вещество способно в одних случаях реагировать как имеющее одно, в других — другое строение, например, как заключающее иногда группу СН2 —СО, иногда ОН = СОН — кето-энольная изомерия или HCN↔HNC. Эти две формы в жидком состоянии или в растворе могут находиться в равновесии. Для продуктов, содержащих вместо водорода «более тяжелые группы (радикалы), могут существовать производные обеих форм: например, RCN нитрилы, RNC изонитрилы (R—углеводородный радикал СН3, C2H5, С6Н5 и т. д.)
Другим обстоятельством, затрудняющим установление строения, является то, что при реакциях нередко имеют место процессы изомеризации, т. е. вместо вещества ожидаемого строения получается, вследствие перегруппировки атомов, вещество иного строения, образование которого трудно было и предвидеть. Это происходит или вследствие того, что в сложном ходе реакции нормального строения вещество и не образуется, или же, хотя оно и образуется, но тотчас переходит в вещество иного строения. Процессы изомеризации могут быть более или менее глубокими. К первым относятся перемещения двойных связей или ненормальное помещение замещающей водород группы или атома, ко вторым — глубокие перегруппировки углеродных цепей или циклов с переходом в циклы мной величины или открытые цепи. Для уяснения процессов изомеризации весьма важны, имеющие и в других отношениях огромное значение, изучение механизмов органических реакций, разложение суммарных процессов на отдельные фазы. Изучение процессов изомеризации нахождение здесь закономерностей, позволяющих предвидеть образование продуктов, образование которых казалось ранее неожиданным и ненормальным, привлекает теперь большое внимание исследователей, и от получаемых здесь результатов можно ждать многого по отношению к выяснению строения изменчивых природных соединений (терпенов, белков) и пониманию механизмов их превращений в живой природе.
Хотя теория строения объясняет громадное число случаев изомерии, но объяснить все случаи изомерии она оказалась неспособной. Потребовалось дальнейшее развитие теории строения, и таким этапом ее развития является стереохимия (см.), которая занимается уже рассмотрением расположения атомов в пространстве.
Итак, в основе современных теорий строения вещества в химии органической лежит понятие об атомности, валентности (Vаlenz) или значности элементов (Wertigkeit). Для химии органической это понятие оказалось особенно плодотворным потому, что валентность углерода в громадном числе соединений неизменно = 4. Притом она одинакова по отношению к водороду и кислороду. Валентность многих других элементов вообще переменна, и валентности по отношению к водороду и кислороду дополняют друг друга до 8. В понятие о валентности и о связи атомов уже вносится свет теориями о строении атомов. Различают два рода валентностей — главные и побочные, и два рода связей — полярную, в электролитах, например NaCl, и типичную неполярную, например в СН4. В первом случае, вследствие отдачи натрием электрона хлору, натрий заряжен положительно, хлор отрицательно, и сила их связывающая — электростатическое притяжение; в случае типичной неполярной связи передача электронов места не имеет, а связью служат при простой связи два общих электрона, при двойной связи — две пары, при тройной — три пары. Между этими двумя типичными видами связи существуют переходы. Неполярным характером объясняется вязкость связи углеродных атомов между собой, обусловливающая возможность сложных углеродных скелетов и большое число изомеров, медленность многих реакций и прочее. Развитие понятий о валентности имеет свою сложную историю, в которой участвовали и участвуют многие выдающиеся химики и физики.
Классификация и номенклатура. При громадном числе органических соединений вопрос о классификации и систематике их имеет особое значение. В основу классификаций обычно кладутся: элементарный состав, число атомов углерода, молекулярный вес, строение (углеродного и другого) скелета, химический характер соединения — степень богатства водородом, присутствие тех или иных групп и особое отношение, которое требует пояснения и носит название гомологии. Так как все соединения, в конечном счете, можно производить от углеводородов, то основными соединениями являются углеводороды. Их, в свою очередь, делят на углеводороды с открытыми цепями (жирного ряда) и циклические; эти последние делятся на углеводороды алициклические, в которых связи и характер близки к углеводородам жирного ряда, и на углеводороды с характером ароматическим. В пределах каждого большого класса (из 3) углеводороды могут быть распределены в гомологические ряды. Состав членов гомологического ряда может быть выражен общей формулой. Общая формула предельных углеводородов — СnН2n+2; остальных рядов — CnН2n, СnН2n-2 и т. д. Во всех углеводородах (за ничтожным числом исключений) число атомов водорода четное. Все члены гомологического ряда обладают сходным химическим характером. Два рядом стоящих члена отличаются на постоянную разность СН2, и физические свойства членов гомологического ряда (при сходстве строения) изменяются закономерно. Отношение гомологии для алкоголей было открыто еще Шилем в 1842 г. Но обобщением, широким применением и самым названием — гомологи, — мы обязаны (1843—1845) известному французскому химику-реформатору Жерару. В гомологические ряды могут быть расположены и разнообразнейшие производные углеводородов — продукты замены в них водорода элементами или сложными группами — радикалами. Радикалы могут быть неорганическими — ОН, NO2, NH2, SO3Н, SH и т. д. или органическими. Среди последних особо важны радикалы углеводородные: СН3 метил, С2Н5 этил, С6Н5 фенил, и кислотные, например, СН3СО ацетил, С6Н5СО бензоил; двухвалентный радикал С=О называется карбонилом; присутствие его характеризует альдегиды, кетоны и кислоты; в первых он входит в виде группы
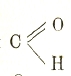
альдегидной, например,
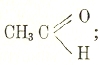
во вторых он связан с двумя углеводородными радикалами, в кислотах в виде группы
- карбоксила.
Продукты замены водорода в углеводородах на ОН представляют разнообразнейшие спирты, или алкоголи. Заменяя, в свою очередь, водород группы ОН в спиртах на радикалы углеводородные, получают простые эфиры, например,
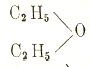
простой этиловой т. н. (неправильно) серный эфир; а на кислотные — сложные эфиры, например,
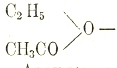
— уксусно-этиловый эфир.
Азотистые соединения тоже очень многочисленны и разнообразны. Многие из них можно рассматривать как производные неорганических соединений азота: азотной и азотистой кислот, аммиака, гидразина или гипотетических соединений, как диимид NH = NH. Соединения, заключающие группу NО2, связанную с углеродом, называются нитросоединениями, а группу NH2 — аминосоединениями. Первые находят применение в качестве взрывчатых веществ и служат переходными звеньями в ряду ароматическом от углеводородов к другим соединениям. Аминопроизводные углеводородов и спиртов обладают основным характером. Особые классы азотистых соединений образуют цианистые соединения, заключающие группу CN, и производные разнообразных азотистых гетероциклов, входящих в состав множества биологически важных соединений. Перечисленные случаи замены водорода различными группами не исчерпывают имеющего место в действительности разнообразия. Дело осложняется еще следующими двумя обстоятельствами: в соединениях могут одновременно присутствовать не одна, а две или несколько замещающих групп, например:
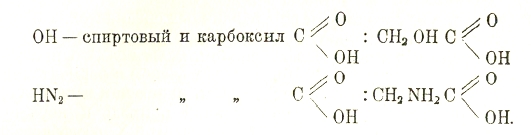
Получаются соединения смешанной функции: спиртокислоты, аминокислоты и т. д. Число различных групп, могущих входить в частицу, может быть значительным, что обусловливает многочисленность, разнообразие и сложность реакций, свойственных соединениям смешанной функции. К подобного рода соединениям относятся, например, углеводы, простейшие из которых, глюкозы, являются в одно и то же время альдегидами или кетонами и многоатомными спиртами. Кроме соединений, строение которых установлено вполне или отчасти, известно много органических соединений еще мало изученных, о которых мы знаем иногда только состав; такие соединения еще не могли занять определенного места в системе и описываются в виде отдельных групп, например, горькие или индифферентные вещества, смолы и прочие природные соединения.
Номенклатура. В настоящее время в химии органической нет единой, общепринятой, рациональной номенклатуры. Наряду с названиями, указывающими, хотя отчасти, на состав и характер соединения (см. выше классификация), существует масса тривиальных названий, даже по отношению к веществам установленного строения, например древесный спирт, различные кислоты, серный эфир, масло горьких миндалей и т. д. В последнее время делаются попытки составить словарь химических тривиальных названий. Попытки выработать рациональную номенклатуру делались. Так, в 1892 г. в Женеве собиралась международная комиссия из известнейших химиков различных стран. Комиссией этой были положены в основу создания номенклатуры следующие принципы, принятые конгрессом (в Женеве):
1) в основу кладутся реакции замещения; 2) название должно указывать на присутствие в различных производных одного семейства общего радикала; 3) функции химических соединений с общим радикалом указываются приставками или окончаниями, добавляемыми к радикалу; 4) названия составляются по формуле и, если присутствует несколько радикалов, их называют раздельно и при указании функциональных групп придерживаются неизменного порядка.
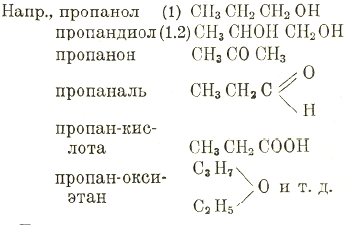
Например, названия предельных углеводородов оканчиваются, как обычно, на ан, этиленовых на ей, ацетиленовых на ин; к циклическим прибавляется цикло:
СН3—СН2—СН3 пропан, СН3СН = СН2 пропен,
Циклопропан. Спиртовая группа ОН указывается приставкой ол, кетонная С=О он, альдегидная аль; место этих групп указывается цифрами, число — приставками к ним ди, три и т. д.
Должно заметить, что женевская номенклатура не вытеснила обычную.
Несмотря на блестящие результаты, достигнутые в химии органической в XIX и первой четверти XX века, здесь предстоит разрешить много важнейших проблем и сделать неожиданных открытий. Здесь, как и в других областях знания, по мере развития науки увеличивается граница, отделяющая известное от неизвестного, и впереди предвидится процесс дальнейшего, неограниченного развития знаний в этой области. С дальнейшим развитием химии органической или родственных наук, методами и результатами которых она пользуется, несомненно, возникнут вопросы и выдвинутся задачи совершенно новые, которые трудно предвидеть. Но и от развития ее по путям, которые можно наметить в настоящее время, несомненно, получатся результаты чрезвычайно важные как для разъяснения загадок строения вещества и внутримолекулярных сил и многих вопросов, связанных с жизнью, так и для дальнейшего развития промышленности и земледелия.
Так, прежде всего, нуждаются в глубоком изучении и уточнении основные химические понятия: о молекуле и природе валентности. Последнее понятие, по-видимому, будет уясняться в связи с учением о строении атомов. В связи с этим находится и вопрос о характере связи между атомами, образующими молекулы, и о работе, необходимой для разрыва связей между различными атомами органических соединений. Новейшие работы по термохимии обещают пролить свет на эти вопросы. Далее, химии органической предстоит:
1. Разъяснить механизмы не разъясненных еще органических реакций, опираясь, где можно, на физическую химию.
2. Пользуясь знанием механизмов реакций, выработать простые, удобные, выгодные технически способы получения органических соединений, играющих важную роль в лабораторной практике, жизни и технике. Примерами подобного рода могут служить получение тростникового сахара из дешевых углеводов, жирных кислот из нефти, синтез хинина и т. д.
3. Выяснить строение сложнейших продуктов жизнедеятельности организмов: сложных углеводов типа крахмала и клетчатки, белковых веществ, сложных терпенов, пигментов, а также продуктов дальнейшего изменения их — гуминовых веществ почвы, играющих весьма важную роль в земледелии, и пр. Заключительным аккордом должен явиться синтез этих соединений в условиях нежных, по возможности близких к природным. В этом отношении чрезвычайно важных результатов следует ожидать от изучения различных органических реакций, протекающих под влиянием катализаторов (см. катализ). Это послужит переходным звеном к ферментативным процессам, играющим такую важную роль в живой природе и изучаемым уже биологической химией, обособившейся в особую дисциплину.
4. Объединить в более общие законы множество частных правильностей, касающихся как физических свойств в связи с составом, молекулярным весом и строением, так и химических реакций.
5. Продолжать разрабатывать вопрос о взаимном влиянии атомов, друг с другом непосредственно не связанных, на химический характер соединений. Вопрос этот, выдвинутый А. М. Бутлеровым, В. В. Марковниковым, Вант-Гоффом и другими учеными, представляет большую важность и обещает привести к общим законам, опираясь на которые можно будет, хотя бы приблизительно, а priori определять свойства и реакции соединений.
6. Систематически и подробно изучить процессы полимеризации и изомерных превращений, имея ввиду добиться общих закономерностей и объяснения причин, почему при известных условиях наступают процессы изомерных превращений.
7. Наконец, следует постепенно подвергнуть критической и, если нужно, новой экспериментальной обработке громадный фактический материал химии органической, чтобы освободить его от сомнительных и даже ошибочных данных.
Литература. А. М. Бутлеров, «Введение к полному изучению органической химии» (посмертное издание). СПБ. 1887. С. Schorlemmer, «Origine et développement de la chemie organique». Paris. 1885. Sir W. Tilden, «Chemical Discovery and Invention in the 20 Century». London. 1916. F. Heinrich, «Theorien der organisсhen Chemie». 5 Auf. 1924. V. V. Richier-R. Anschütz, «Chem. der Kohlenstoffverbindungen», I и II т. 1910—1913. I. Schmidt, «Kurzes Lehrb. der organ. Chem.» 1922. V. Meyer and С. Jacobson, «Lehrb. der organ. Ch.», 6 т. Руководство по химии органической: Н. Я. Демьянова, В. Н. Ипатьева, С. Н. Реформатского, А. Н. Реформатского, Д. Е. Фаворского, А. Е. Чичибабина и др.
Н. Демьянов.
| Номер тома | 45 (часть 2) |
| Номер (-а) страницы | 348 |