Селитры
Селитры — соли азотной кислоты щелочных» (калия, натрия и др.) и щелочноземельных (магния, кальция, стронция, бария) металлов. Под именем просто селитры чаще всего разумеют азотнокислый калий, KNO2, а чилийской селитра — азотнокислый натрий NaNO2. Первая имеет большое применение в пороходелии, вторая — для удобрения и приготовления азотной кислоты.
Селитра образуется в природе при разложении азотистых органических веществ в присутствии щелочей идя щелочных земель, при большом доступе воздуха и влаги, при температуре около 37°С. Поэтому в теплых странах процессы селитрообразования идут более успешно; например, в Индии (Бенгалии) богатые запасы селитры, извлекаются из почвы. Селитросодержащая почва в наступающее после дождей жаркое время покрывается налетом селитры. Эти выветрелости собирают, методически выщелачивают и подвергают увариванию.
В течение долгого времени, вплоть до появления на рынке чилийской селитры, калийная селитра готовилась во всех государствах Европы искусственным путем в так называемых селитриницах. По Вагнеру добывайте в селитриницах ведется следующим образом. Выбирают землю, богатую углокальциевой солью (мергель), мусор строений, золу: древесную, торфяную, каменного или бурого угля, дорожную пыль и грязь, городские и конюшенные отбросы, или из прудов и шлюз, известковые остатки содовых, стеклянных, бумажных, белильных заводов, золу и известь с мыловарен, в затем, как источники азота: навоз, животные отбросы, мясо, жилы, кожу, кровь с скотобоен, остатки кожевенных и клееваренных заводов, шерстяные тряпки с ткацких заводов и т. д., к свежим животным отбросам примешивают части растений, особенно из числа содержащих селитру — ботву картофеля, свеклы, крапиву, подсолнечник, огуречник и т. п. и поливают кучи время от времени навозной жижей. Из этой смеси устраивают на твердой почве, лучше всего на утрамбованной глине, кучи вышиной в 2—2,5 метра, формы притупленной пирамиды с прослойками из соломы и хвороста, для возможно большего доступа воздуха со всех сторон. Можно также производить гниение животных веществ в отдельных кучах и затем только смешивать их с остальными веществами в больших кучах. Когда процесс образования селитры в такой селитрянице приближается к концу, ей дают обсыхать: на поверхности земли ее образуется кора толщиной в 6—10 см, более богатая солями азотной кислоты, чем остальная масса, так как действием капиллярности щелок собирается на поверхности и здесь испаряется. Эта кора постоянно снимается, пока, наконец, вследствие недостаточного притока воздуха к внутренним частям, образование селитры не прекращается: тогда кучу обкладывают выщелоченной уже землей или разрывают ее и переделывают заново. В некоторых кантонах Швейцарии селитру добывают на скотных дворах, построенных на склонах гор. Под хлевом делают яму глубиной 0,6—1,0 метра, засыпают ее пористой землей, содержащей необходимое количество извести, и утаптывают ее. По прошествии 2—3 лет снимают во время отсутствия скота досчатую настилку, выгребают селитряную землю и выщелачивают ее водой. Раствор выпаривают в сарае, в обыкновенном котле, вкопанном в землю над ямой. Выщелоченная земля всыпается обратно в хлевы. Вторично выщелачивать землю из того же хлева можно только по прошествии нескольких лет. Один хлев дает, говорят, от 25 до 100 кг сырой селитры, по другим сведениям, даже до 500 кг. Раствор смешивается с золой и известью, сливается с осадка, выпаривается и подвергается кристаллизации. Выделившиеся кристаллы помещают сначала в корзины для стока оставшегося раствора, вкладывают в мешки и отправляют в рафинировачные отделения при пороховых заводах.
Добывание селитры в селитриницах или буртах прежде доставляло главную массу селитры в Европе, но теперь утратило свое значение. До ХIХ в. все европейские государства сами добывали для себя селитру из туземных материалов, причем во многих государствах доставка ее из материалов для ее получения представляла натуральную повинность населения, например, в некоторых местах Франции, в Швеции, в древней России, фабриканты очищенной селитрой пользовались покровительством властей, обеспечивавших им большие привилегии. Народу было повелено оказывать им во всем содействие и под страхом крупного штрафа воздерживаться от выкапывания селитры в своих собственных или других домах, конюшнях, хлевах и т. д. Сени и конюшни запрещалось вымащивать камнями, а где это было, все замощенное вырывалось. Горных и послушных подданных усовещивали с терпением переносить эту неприятность, как необходимую для защиты страны и парода, ввиду потребности для армии иметь достаточный запас пороха. Тем не менее привилегии эти постоянно вызывали недовольство, переходившее во многих случаях в смуту.
К XVIII в. стали известны богатые месторождения селитры в Европе, Индии и Америке. В этом веке, по крайней мере, одна треть юго-западных областей Испании оставалась неразработанной и служила исключительно для добывания селитры из верхнего слоя почвы, который и выщелачивали в добытую кальциевую селитру кипятили с раствором щелока, калий которого замещал собою кальций. Таким способом получали хорошую селитру. Большие залежи селитры были открыты в начале XVIII в. в Индостане, особенно на Цейлоне и в Бенгалии. На Цейлоне сырая селитра была найдена в скалистых пещерах, служивших, вероятно, раньше жилищем зверям и человеку, а теперь сделавшихся приютом бесчисленному множеству летучих мышей, трупы и помет которых послужили для нее источником азота.
В Бенгалии она находится в верхнем слое почвы, богатой органическими составными частями, где условия благоприятствуют образованию значительных количеств ее. К началу сухого времени года селитра помощью волосности поднимается на поверхность почвы и собирается там в виде корки.
В XIX в. начала привозить в Европу индийскую селитру, и она стала заменять часть туземной, но значение ее еще было второстепенно. Только в XIX в., около 1815 г., привоз индийской селитры настолько усилился, что под влиянием его производство туземной селитры в Западной Европе стал приходить в упадок. Еще более сильный удар этому производству был нанесен возникновением переработки американской натровой селитры в калийную, что произошло в 50-х гг., во время Крымской войны. Еще в 40-х гг. наибольшее количество азотнокислого калия для пороха во всей Европе добывалось буртовым способом; в 50-х гг. буртовой способ еще поддерживался правительством Западной Европы, но в 70-х гг. был там уже оставлен.
В России добывание селитры из буртов, т. е. куч препарованной селитряной земли, начато было при Иване Грозном, причем доставка некоторого количества селитры составляла натуральную повинность, например, в Новгороде. Так как туземной селитры не хватало на военные надобности, а между тем иностранцы не считали благоразумным продавать России этот товар, то при царе Федоре Ивановиче было сделано распоряжение, чтобы воск, которые иностранцы очень ценили, продавали не иначе как на селитру. Другое указание на бедность тогдашней России селитрой мы находим в описании царствования Алексея Михайловича: при описании приема персидских послов боярами упоминается, что бояре заявили желание получать селитру из Персии. В последующее время было достаточно организовано получение в России туземной селитры. В XVIII и XIX вв. буртовая селитра добывалась частью в Малороссии, частью в Поволжье. В Малороссии добывали селитру еще во время гетманов. В Приволжском крае селитряные заводы были основаны Петром Великим, а именно, один в селении Болгары в Спасском уезде Казанской губернии, а другой близи Астрахани, но заводы эти недолго существовали. Петром Великим был построен селитряный завод близ Саратова во время его дохода на Азов. В Спаском уезде, Казанской губернии, в месте, называемом Селитряный овраг, найдены были в 60-х гг. XIX в. заброшенные бурты, давно поставленные, а ниже Царева на Ахтубе есть место, называемое Селитряный городок. Производство селитры в приволжских губерниях было возобновлено с конца 50-х гг. XIX в. В обеих местностях, как уже было выше замечено, селитряный промысел пришел теперь в совершенный упадок. Несмотря на дороговизну буртовой селитры в сравнении с конверсионной, наше правительство долго поддерживало искусственно буртовой промысел, в видах возможности европейской войны и запирания нашей границы. Но во 2-й половине 80-х гг. эта поддержка была оставлена. К этим историческим данным, заимствованным нами у профессора П. Н. Любавина (см. «Техническая химия», т. II, глава III-я), добавим, что в 1858 г. военным ведомствам для распространения у нас селитряной промышленности предложено было составить два руководства по селитряному делопроизводству — одно для заводчиков, другое для крестьян, вследствие чего член артиллерийского отделения профессор химии Ходнев командирован был в Малороссию с целью ознакомиться на месте со средствами селитропроизводительных губерний, изучить в подробности тамошние способы выварки селитры и потом, основываясь на собранных данных, составить требуемые руководства для помещиков и крестьян. По исполнении этого поручения профессор Ходнев представил составленное им «Руководство к селитроварению в Малороссии и в прилежащих к ней селитропроизводительных губерниях». Артиллерийское отделение признало полезным его напечатать на казенный счет в числе 1 200 экземпляров и стараться о сколь возможно большем распространении у нас этого сочинения (см. Артиллерийский журнал, 1858 г.). В статье проф. Ходнева «Селитряный промысел в России» (см. Артиллерийский журнал, 1858, стр. 91—128) указывается, что в 1844 г. Вольно-Экономическое Общество, желая способствовать улучшению нашего селитроварения, полагало полезным учредить школу для образования опытных мастеров-селитроваров. «Но благая мера эта осталась неосуществленной». Далее приводятся данные о количестве добываемой в различные года селитры; так, с 1854 г. помещикам всех селитропроизводительных губерний было предоставлено поставлять столько селитры в казну, сколько они могут. Но, несмотря на все меры, принятия помещиками (бурты давно заброшенные, старые навозные залежи, бурты недавно непереработанные все пошло в дело, и возникло много новых заводов), можно было получить в год не более 200 тысяч пудов, как это показывают официальные данные о поставках на Шостенский пороховой завод: 1854 г. — 128 838 пуд., в 1855 г. — 220 454, в 1856 г. 203 587, в 1857 (по 1-е ноября) — 195 447 пуд. С развитием добывания селитры в Чили и ввоза ее в Европу и увеличением потребности в навозе для целей удобрения, производство селитры в буртах начало постепенно падать и к концу ХІХ-го века почти совершенно прекратилось. Калийную же селитру начали приготовлять из чилийской. Для этого пользуются поташом (K2CO3), обыкновенно же хлористым калием KСІ. При употреблении поташа к горячему концентрированному раствору последнего прибавляют эквивалентное количество горячего же крепкого раствора чилийской селитры; при нечистых материалах пропорция смешиваемых солей должна быть установлена по действительному содержанию в них K2CO3 и NaNO3. Тотчас наступает обменное разложение, по уравнению: K2CO3 + 2NaNO3 = 2KNO3 + Na2CO3, причем в растворе остается KNO3, сода же Na2CO3 в безводном состоянии осаждается частью тотчас, частью при дальнейшем уваривании. При охлаждении раствора кристаллизуется селитра, которая, однако, содержит примесь соды и должна быть очищена вторичной кристаллизацией. Удерживающаяся выделявшейся содой селитра удаляется промывкой насыщенным раствором соды. В настоящее время для обменного разложения пользуются почти исключительно Стассфуртским хлористым калием. Азотнокислые соли калия и натрия в горячей воде растворимы гораздо более, чем в холодной: растворимость же хлористых соединений, особенно хлористого натрия NaCl, с повышением температуры изменяется весьма мало. 100 частей воды растворяют при 0°С 35,52 части, а при 100° — 40,35 частей хлористого натрия (NaCl), азотнокислого же калия (KNO3) при 0°13,°3 ч., а при 100° — 301,0 час. При смешении хлористого калия, КСІ, и азотнонатровой соли происходит реакция двойного обмена по уравнению KCl + NaNO3 = KNO3 + NaCl. Если водный раствор настолько разведен, что все получающиеся соли остаются в растворе, то в нем находятся четыре соли, ибо не все частицы хлористого калия могут перейти в частицы азотнокислого кальция, так как последние, встречаясь с частицами хлористого натрия, могут вступать с ними в реакцию двойного обмена, по уравнению: KNO2 + NaCl = KCl + NaNO3. Если же мы начнем выпаривать раствор, то хлористый натрий (растворяющийся в горячей воде почти в такой же степени, что в холодной) будет оседать из раствора и уходить таким образом из сферы действия солей, находящихся в растворе, вследствие чего могут образоваться путем обменного разложения между хлористым калием и азотнокислым натрием новые количества калийной селитры и хлористого натрия. Сливая горячий раствор с осевшего хлористого натрия и охлаждая его, получают кристаллы азотнокислого калия (ибо он менее растворим в холодной воде, чем в горячей) с небольшой примесью хлористого натрия, от которого очищают новой кристаллизацией и литрованием. Литрованием называют процесс промывания кристаллов селитры измельченной в виде муки насыщенным раствором селитры; такой раствор не способен растворять селитру, а растворяет только примеси. Азотнокислый калий KNO3 кристаллизуется в виде бесцветных длинных, по бокам бородчатых ромбических шестигранных призм, оканчивающихся такими же пирамидами. Кристаллы удельного веса 2,058 плавятся при 329° в бесцветную жидкость; при температуре красного каления селитра разлагается на азотистокислую соль и кислород. Селитра нерастворима в абсолютном спирте; в воде растворяется с заметным понижением температуры. Насыщенный раствор кипит при 118°С. При возвышенной температуре селитра действует как сильное окисляющее средство, сжигает органические вещества, уголь, серу и т. п. Главнейшее применение калийной селитры находит при производстве черного пороха и в пиротехнике. В смеси с поташом употребляется в металлургии черный и белый плавень. В смеси с бурой применяется для плавки благородных металлов. В небольшом количестве употребляется при солке мяса для придания последнему красного цвета, хотя для этой цели вполне пригодна и чилийская селитра.
Чилийская селитра или азотнокислый натрий (NaNО3) добывается в Чили, где очень значительные залежи находятся на западном берегу Южной Америки между 19° и 25° градусами южной широты на пространстве около 1000 километров сухой и лишенной растительности пустыни. Эти селитряные поля расположены в северных провинциях Чили: Тарапака, Токопилла, Антофагоста, Талтал, Атакама не на самом берегу океана, но на расстоянии от него 25—30 миль, будучи отделены от него невысоким горным кряжем, идущим параллельно Андам и от них независимым, — так называемым Береговым Кордильерам. Залежи не занимают сплошь всего указанного пространства, но с перерывами, накопляясь преимущественно в углублениях почвы, вблизи горного кряжа. Чилийская селитра обязана своим происхождением гниению давно уже погибших организмов. Мнения о причинах скопления в одном месте чудовищных масс таких остатков все еще расходятся. Пространство, на котором находили селитроносную породу, представляло в 60-х годах 1 164 миллиона кв. метров. По другим указаниям залежи натровой селитры занимают площадь в 120 миль в длину и 2 мили в ширину, или 4 756 миллионов кв. метр. С 1 кв. версты площади получается средним числом 1 центнер селитры, но иногда добыча доходит до 24 цент. Азотнокислый натр в более или менее чистом виде встречается здесь редко, представляя иногда белые гнезда; обыкновенно же он сопровождается большим количеством хлористого натрия и другими солями селитры растворимыми в воде, а также песком и глиной. Эта селитроносная порода, представляющая конгломерат песка и глины и называемая калише (caliche), составляет пласт от ¼ до 1,5 метра толщиной и лишь в редких случаях выступает на поверхность. В большинстве же случаев калише залегает под наносными глыбами «костры», мощностью в среднем 50 см.
Калише бывает обыкновенно буро-серого цвета и грубо-зернисто, реже встречается бурое мелкозернистое, еще реже желтое и белое; встречается тоже красное, черное, синее, полосатое и бесцветное калише. Чем больше содержание азотнокислого натра, тем белее порода. Калише иногда твердо и с ясно выраженной кристаллизацией, иногда рыхло и мягко; более чистое отличается твердостью и плотностью. Иногда оно столь твердо, что рвут порохом. Содержание азотнокислого натра в нем представляет большие колебания, по одним от 20% до 64%, по другим — от 30% до 80%. Для добывания азотнокислого натра куски породы сортируют руками, очищая от землистых частей, и затем вываривают с водой, причем отделяют селитру от примешанной к ней поваренной соли. Для этого пользуются неодинаковой растворимостью в горячей и холодной воде; при 0° в 100 частях воды растворяется 73 части NaNO3, а при 100°С 200 ч. (о растворимости хлористого натрия см. выше). Способ работы, применявшейся прежде, был чрезвычайно груб, но теперь его значительно улучшили.
В 60-х годах перерабатывали лишь руду с содержанием не менее 50% NaNO3. Калише, содержавшие менее 40% NaNO3, не идет в работу, а отбрасывались; кроме того извлечение азотнокислого натра производилось на месте очень несовершенно, так что из руды с 65% NaNO3 получало не более 34%—З5% NaNO3. Вследствие этого там накопились отвалы, содержащие еще много селитры, за разработку коих принялись впоследствии, когда цены на селитру поднялись, и, кроме того, выступал новый конкурент — Потоденская селитра из воздуха (см. ниже). В настоящее время обрабатывают калише с 18—20% селитры. Для этого измельченная машинами сырая селитра помещается в особые сосуды для выщелачивания, которые составляют целый ряд соединенных друг с другом железных ящиков, поставленных таком образом, что из одного ящика раствор может спускаться в другой. Нагревание производится помощью закрытых паровых труб, в которые пускают пар под давлением 4—5 атмосфер. Вода, конденсирующаяся в этих трубах, стекает обратно в паровой котел, что при недостатке воды в тех местностях является весьма важным обстоятельством. По окончании растворения, горячий насыщенный раствор сырой селитры спускают в кристаллизационные ящики. Не растворившиеся остатки, содержащие не более 4—5% селитры, автоматически удаляются через особые отверстия, устроенные на дне ящиков, откуда они попадают прямо в вагонетки и отвозятся прочь. По мере охлаждения раствора, селитра выкристаллизовывается, тогда как поваренная соль остается в растворе. По истечении 4—6 дней, кристаллизация оканчивается, кристаллы вынимают и кладут на особо устроенную наклонную поверхность, чтобы дать возможность стечь маточному раствору, после чего кристаллы селитры промывают помощью лейки с ситом для удаления захваченного ими раствора поваренном соли. Полученную таким способом селитру сушат на открытом воздухе и затем упаковывают. В торговлю поступает чилийская натровая селитра в виде мелких зерен величиной с горошину или чечевицу, кубической внешности, буроватого цвета, обыкновенно несколько влажных, с содержанием 95—98% NaNО3.
Разработка залежей селитры началась в тридцатых годах прошлого столетия, и с тех пор она увеличивалась с каждым годом, особенно же с 1860 г., когда чилийская селитра начала применяться для нужд сельского хозяйства: вывоз ее равнялся в 1825 г. 935 тоннам, в 1835 г. — 7 020 т., в 1845 г. — 18 814 т., в 1860 — 68 512 т., в 1870 — 147 170 т., в 1880 — 226000 т., в 1890 — 1 065 000 т., в 1900 — 1 460 000 г., в 1908 г. – 2 014 000 т., в 1910 — 2 308 000 т., причем в 1910 г. в Европу было ввезено 1 651 000 т., в Соединенные Штаты – 565 000 т., в другие страны — 92 000 т. Потребление же чилийской селитры в 1910 г. в Европейских странах равнялось:
В Германии 709 000 т.
Франции 336 000
Бельгии 262 000
Голландии 132 000
Англии 120 000
Италии 38600
Испании 6480
Австрии 5 600
Швеции 2000
ВСЕГО 1611000
Что касается России, то в 1908 г. оно равнялось 838 228 пуд. или около 14000 т. Профессор Юриш (Jurisch) вычислив, что в 1905 г. потребление чилийской селитры приходилось на одну голову жителя
в Бельгии 44 фунта
Германии 19
Голландии 14,7
Франции 12,7
Дании 10,4
Швеции 9,5
Соединенные Штаты 7,3
Англии 5,0
Австро-Венгрии 3,1
Италии 3,0
Швейцарии 1,4
Норвегии 0,5
Быстрое увеличение потребления селитры поставило на очередь вопрос о времени истощения ее запасов. Ответ на него является нерешенным, так как о количестве находящейся в залежах имеются разноречивые показания.
Так оно равняется вычислениям
Смита в 1860 г. 63 000 000 тонн
Даранского в 1887 г. 89 050 000 т
Лагранжа и Экманна в 1892 г. 160 000 000 т
Германского Консула в 1900 г. 65 000 000 т
Чилийского общества пропаганды селитры в 1910 г. 246 000 000 т.
Точно так же для срока истощения запасов получаются разнообразные числа: в 1888 г. чилийское правительство определило этот срок до 1913 г., в 1909 г. до 1940, а в 1907 г. до 2032 г.
Разнообразие этих цифр объясняется, во-первых, открытием новых залежей, а во-вторых различием гипотетических оснований, служивших для вычислений.
Разработка залежей чилийской селитры принадлежит большому числу компаний: в 1905 г. общий капитал 62 компаний равнялся 112 000 000 долларов. Число отдельных предприятий возросло в 1911 г. до 158. Общая сумма капиталов всех этих предприятий достигла 136 500 000 долларов, причем, английских 53 500 000 дол., чилийских 51 500 000 дол., германских 16 500 000 долларов и других 35 000 000 дол.
В течение 31 года, начиная с 1879 г., промышленники и агрономы всего мира уплатили чилийской промышленности 425 000 000 долларов.
Синтетическое получение азотной кислоты и ее солей из воздуха. Непрерывно возрастающее потребление заставало многих задуматься над вопросом о том, как предотвратить грозящее истощение почвы вследствие недостатка такого питательного вещества, каким является чилийская селитра. Еще в 1898 г. известный английский ученый Крукс указал, что вопрос о синтетическом добывании азотной кислоты и ее солей из азота и воздуха является вопросом жизни и смерти для будущих поколений.
Но тот же Крукс, нарисовав мрачную картину грядущего голода, указал на то, что лабораторные следования укажут способ, а гидравлические силы, которыми сравнительно мало пользовались, позволят человечеству применить этот способ, чтобы спастись от голода. Предсказания английского ученого сбылись: мы владеем несколькими техническими способами для утилизации азота воздуха, одним из коих является получение при помощи электрической искры окислов азота. Еще г. 1781 г. Кавендиш сжигал водород в избытке воздуха и нашел, что вода образовавшаяся при этом, содержала азотную кислоту, а в 1784 г. он заметил, что при пропускании электрических искр через воздух образуются окислы азота, поглощаемые раствором едкой щелочи. В 1789 г. Луи Одьер в «journal de Chimie et de Physique» сообщил об образовании окислов азота из кислорода и воздуха при высокой температуре. Первый патент на производство азотной кислоты из воздуха был взят в Англии в 1859 госпожой Лефебр (из Парижа). Прибор ее представлял стеклянный баллон, снабженный четырьмя трубками; через две из них проходят два электрода с платиновыми наконечниками, между коими проскакивала искры. Через третью пропускался воздух, а четвертая соединялась с поглотительными сосудами, наполненными водой. Г-жа Лефебр указала на то, что при избытке кислорода выходы окислов азота увеличивается. Затем до конца ХІХ в. появился еще ряд патентов, не имевших практического значения. В 1902 г. основалась «Atmospheric Products Со», которая, пользуясь силой Ниагарского водопада, применила патент Р. Ловджой и Э. Бредлей, для получения окислов азота с помощью тонких и длинных электрических искр. Постоянный ток получатся от особой машины, дающей непосредственно напряжение в 15 000 вольт. В течение часа в одном аппарате перерабатывалось 11 ½ куб. метров (или около 34,5 кг) воздуха, в выходящих газах находилось 2 ½ % окиси азота, что при пересчете на азотную кислоту составляло 83 грамма на 1 киловатт-час. Такой выдающийся результат удалось получить впервые, но успех оказался обманчивым, и летом 1904 г. компания прекратила производство. В 1903 г. Христиану Биркеланду (профессору физики в Христиании) в сотрудничестве с инженером Самуэлем Эйде удалось найти технический способ получения окислов азота, оказавшийся выгодным.
К этому же времени благодаря трудам Нернста, Габера и др. выяснялись химико-физические условия образования окислов азота. Нернст показал, что при действии не только электрической искры, но и вообще высокой температуре азот соединяется с кислородом и образует окись азота NO, по уравнению: N2 + O2 = 2NO.
Но эта реакция, как большинство реакций в газообразной среде, обратима, т. е. при той же температуре окись азота может распадаться на кислород и азот: 2NO = N2 + O2. Поэтому образование окиси азота ограничено пределом, т. е. не вся смесь кислорода и азота, взятых в равных объемах, может превратиться в окись азота, но при данных условиях температуры в давлении реакция останавливается, когда содержание окиси азота достигает известной величины. Предел наступает тогда, когда в каждый момент образуется столько же молекул, сколько распадается, что может быть выражено следующим уравнением: N2 + O2 ↔ 2NO. Чем выше температура, тем предел образования окиси азота больше: при 1538°С он равен 0,37%, а при 2927°С он достигает 5,0%. Кроме того, время, в которое эта пределы достигаются, тем короче, чем выше температура; так при 1225°С потребуется более суток, чтобы реакция достигла половины предела, а при 2000°С это достигается в доли секунды.
Отсюда видно, что высокая температура оказывается вдвойне выгодной: во-первых, она повышает процент образующейся окиси азота, а во-вторых, превращение в окись азота происходит быстрее.
Один каловатт*)-год (365х24 часов) теоретически может уловить количество азота, соответствующее 1850 кило азотной кислоты (HNO3), если дуга работает при 4000°С и только 819 кило при дуге, работающей при 3000°С, т. е. падение температуры на 1000° понижает выход более, чем на 50%.
Окись азота, образовавшаяся при высокой температуре, при медленном охлаждении постепенно распадается на кислород и азот. Если реакция образования окиси азота происходит при 2927°С, то содержание окиси азота (NО) достигает 5% (по объему), и затем при медленном охлаждении до 1538°С равновесие устанавливается на пределе, соответствующем этой температуре, т. н. 0,37% т. е. во время охлаждения теряется более 90% того, что было добыто при 2927°С.
Если образование окиси азота происходит быстрее при высокой температуре, то и обратное распадение совершается тем скорее, чем выше температура: иными словами, наиболее опасной для существования окиси азота является температура, которая лежит наиболее близко к температуре ее образования. Чем ниже температура, тем окись азота более стойка. Отсюда прямой вывод: необходимо как можно быстрее охлаждать газы, которые были нагреты до температуры вольтовой дуги для того, чтобы уменьшить возможность распадения образовавшейся окиси азота. Эти условия соблюдаются при получении пламени азота в электрической дуге высокого напряжения.
Согласно Нернсту образование окиси азота в вольтовой дуге обусловливается исключительно ее термическим действием: дуга является источником высокой температуры, необходимой для распада молекул азота (N2) и кислорода (02).
Но существуют много фактов, которые указывают, что дело не так просто, и что в рассматриваемых явлениях электрический разряд не остается без влияния. Опыты Варбурга, Бертело, особенно же Габера и Кенига позволяют предполагать влияние самого разряда электрической дуги на образование и выход окиси азота. Согласно представлениям, выработанным новой физикой, относительно прохождения электричества через газы как при тихом разряде и токе через сияние, так и при разряде через вольтову дугу от катода (т. е. места выхода отрицательного тока) из твердого или жидкого проводника, в газ отскакивает большое число электронов (т. е. элементарных частиц отрицательного электричества), которые ионизируют своими толчками нейтральные газовые молекулы, т. е. превращают их в электрически заряженные осколки. Эти толчки электронов являются источником энергии, нужной для эндотермической реакции. Габер и Кениг достигли такой большой концентрации окиси азота в световой дуге, что объяснение этого результата чисто теоретическими явлениями может быть допущено со слишком большими трудностями.
*) 1 киловатт равняется 1,36 л. с.
Употребляя короткие пламенные дуги, Габер констатировал весьма высокое процентное содержание образующейся окиси азота, именно 7 —10%. Такая концентрация, согласно опытам Нернста, должна отвечать температуре равновесия около 4300°, тогда как температура дуги при исследованиях Габера во всяком случае ниже 3000°, при которой процент окиси азота достигает всего 3,57%. Несоответствие между опытом Габера и термической теорией выступает вполне ясно и может быть устранено только при допущении участия самого разряда в процессе образовании окиси азота.
На основании своих опытов Габер приходит к выводу, что электрический разряд пламенной дуги является главнейшей причиной образования окиси азота, что, напротив, слишком высокая температура вызывает более или менее значительный распад окиси азота, возникшей под влиянием разряда. Для возможно большего выхода окиси азота следует охлаждать пламенную дугу, чтобы уменьшить разложение.
Какие бы причины не вызывали образование окиси азота в вольтовой дуге, для большего ее выхода нужно, чтобы воздух, прошедший через нее, быстрее затем мог охладиться. Это и достигается в печи Биркеланда и Эйде. Основанием их способа послужил следующий опыт Бкркеланда: он поместил два электрода перпендикулярно к линии, соединяющей два полюса сильного электромагнита (25000 силовых линий), причем расстояние между электродами равнялось 2 мм. Когда был пропущен постоянный ток силой в 2 ампера от динамо-машины в 8000 вольт напряжения, то получился светящийся полукруг и послышался сильный шум наподобие свиста (см. рис. 1).
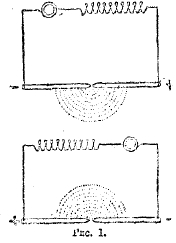
Рис. 1.
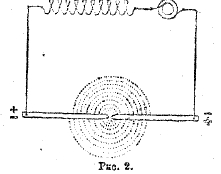
Рис. 2.
Изменяя силу электромагнита, можно изменять высоту тона свиста: чем сильнее электромагнит, тем выше звук. Если пропустить переменный ток высокого напряжения, то пламя получается в виде тонкого диска, причем звук теряет характер свистка (см. рис. 2), Вышеописанные опыты Бвркэланда послужили основанием для технического способа получения окислов азота, разработанного совместно Биркеландом и Эйде.
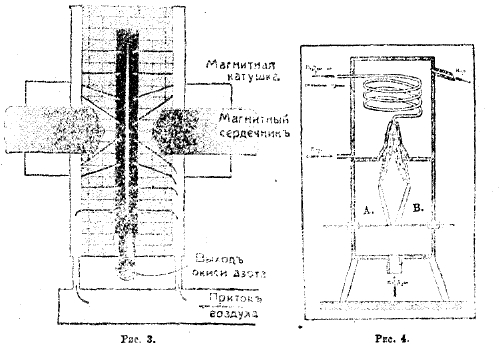
Рис. 3. Рис. 4.
Рис. 3 представляет схематически устройство одной из печей Биркеланда и Эйде. Печь делается из железа, имеет в большинстве случаев форму плоского цилиндра, внутри покрытого шамотом. Выход азотной кислоты достигает максимум 60 граммов киловатт-час. Опыт показал, что пламя, получаемое в печи такой системы, обладает большой стойкостью. С помощью только одной пары электродов, представляющих две медных трубы в 1,5 см диаметром, охлаждаемых внутри током холодной воды и отстоящих друг от друга на 2 миллиметра, получается пламя в виде диска в 1,8 метра диаметром. Эго пламя находится в центральном пространстве печи, имеющем всего только 8 сантиметров ширины, вся же печь высотой в 2 метра построена из огнеупорных материалов, скрепленных металлической обшивкой.
Первая печь была построена в июле 1903 года в Христиании на 6—4 киловатта, или 5 лошадиных сил. Она питалась переменным током в 5000 вольт при 50 периодах в секунду, но уже в сентябре 1903 г. была построена новая печь на 10 киловатт, а затем опыты были перенесены в Vasmoёn, около Арендаля, где можно было пользоваться более могучим источником электричества, и, наконец, в Нотодден. Воздух, который служит для питания пламени, проникает сквозь боковые стенки, построенные из огнеупорного материала. Магнитное поле производится двумя большими электромагнитами, полюсы которых расположены с обеих сторон пламенной камеры и отстоят друг от друга на 10 сантиметров.
Опыты показали, что в печи Биркеланда и Эйде техническим путем почти достигнут предел возможного теоретического получения окиси азота. Газы, выходящие из печи, содержат 1—1 ½ % окиси азота, что составит на 1 киловатт-год (365х24 ч.) 500—600 кг азотной кислоты.
Успех печи Биркеланда и Эйде привлек внимание химиков и техников к этому вопросу, и в настоящее время взято много патентов на устройство печей для сожигания азота воздуха, причем оказалось, что можно получать хорошие выходы окиси азота в печах, действующих без участия магнитного поля. Подобного рода печи были предложены Паулингом, Ги, Шеихерром (Баденской анилиновой и содовой фабрикой), Горбовым и Миткевичем и др.
Весьма проста конструкция печи А. И. Горбова и В. Ф. Миткевича. Между двумя полюсами (или тремя, если пользоваться трехфазным током) образуется пламенная дуга (рис. 4), которая током нагнетаемого воздуха раздувается и приобретает форму, показанную на рисунке. Верхняя часть дуга непосредственно соединяется с холодильником, охлаждаемым током воды. Печь Горбова и Миткевича была испытана ими в электротехнической лаборатории Петербургского Политехнического Института сперва в меньших размерах, а затем на Сестрорецком ружейном заводе в размерах, приближающихся к заводским, а именно — на 250—300 киловатт. Опыты показали, что она позволяет получать в среднем 68 граммов азотной кислоты на каждый кВт-час, горящий в вольтовом дуге, при среднем содержании 2,15% (объемных) окиси азота и выходящих из печи газах. В подобного рода печи в течение года, считая мощность дуги в 186 киловатт и 8500 рабочих часов (вместо 8760), можно получить около 6550 пудов азотной кислоты.
Прежде чем переходить к рассмотрению дальнейших процессов при помощи коих можно перейти от окиси азота до азотной кислоты, укажем, что на основании теоретических расчетов, основанных на том, что граммомолекула (30 г) окиси азота требует для своего образования 21000 калорий, приходят к выводу, что для получения кг азотной кислоты нужно затратить приблизительно 7600000 калорий, или 8,85 кВт-часов. Отсюда теоретически кВт-час (1,36 л. с.) затраченной электрической энергии дает при вделанных предположениях 113 г (100%) азотной кислоты. Из вычислений видно, что главная часть этой энергии, тратится непроизводительно на нагревание массы воздуха. Поэтому повышение процентного содержания окиси азота увеличивает выход азотной кислоты. На практике выходы получаются значительно меньше и обычно не превышают 70 граммов, т. е. около двух третей теоретического выхода.
Образование азотной кислоты из окиси азота происходит не прямо, а через ряд превращений. При высокой температуре окись азота не соединяется с кислородом, но когда температура падает до 620°, то происходит образование двуокиси азота по следующему уравнению: 2NO + O2 ↔ 2NO2. Когда температура воздуха с окислами азота достигнет приблизительно 150°, то вся окись азота, получившаяся в пламенной дуге, превращается в азотноватую окись NO2. Ниже 150° двуокись подвергается дальнейшему превращению по формуле: 2NO2 ↔ N2O2, в результате моего образуется известное количество азотноватого ангидрида N2O2. Таким образом, при обыкновенной температуре получается смесь азотноватой окиси (NO2) и азотноватого ангидрида (N2O4).
При пропускании такой смеси газов в воду получается как азотная, так и азотистая кислоты:
2NO2 + H2O = HNO2 + HNO2
N2O4 + H2O = HNO3 + HNO2
Но тогда как азотная кислота остается без изменения, азотистая кислота вследствие своей непрочности легко распадается с образованием азотной и окиси азота:
3HNO2 = HNO3 + H2O + 2NO
Таким образом, из трех молекул азотистой кислоты только одна идет на образование азотной, тогда как две освобождаются в виде окиси азота. Но в присутствии свободного кислорода выделившаяся окись легко переходит в азотноватый ангидрид, который опять реагирует с водой и т. д. И при избытке кислорода, в конце концов, вся смесь азотноватой окиси и азотноватого ангидрида может быть превращена в азотную кислоту. Такой процесс и происходит при техническом получении последней.
Поглощение азотноватой окиси и азотноватого ангидрида водой протекает до известного предела. Когда концентрация азотной кислоты в растворе достигнет приблизительно 65 %, то дальнейшее поглощение окислов уже прекращается.

Рис. 5.
Технически получается даже не 65%-ная азотная кислота: концентрация технически получаемой «воздушной» азотной кислоты не превышают 50%. Для получения более крепкой кислоты подвергают ее перегонке или другим процессам.
В присутствии озона поглощение окислов азота можно вести значительно дальше и достигнуть весьма высоких концентрации азотной кислоты — до 90%.
Способ помещения окислов азота водой носит название кислого. При щелочном же способе поглощения окислы азота проводят в растворы щелочей или их углекислых солей, а также в башни, в коих находится мел и т. п.
Приготовление азотной кислоты и ее солей из азота воздуха происходит в настоящее время (1917 г.) во многих местах Европы: в Германии, Франции, Швейцарии, но наиболее крупные заводы находятся в Норвегии, где с начала ХХ-го в. в глухой местности Ноттоден создались крупные заводы, произведшие переворот в селитряной промышленности.
Для того, чтобы получать азотистую кислоту из воздуха, нужна, энергия, и стоимость ее определяет издержки производства, а, следовательно, возможность или невозможность строить заводы. В наиболее благоприятных условиях в этом отношении находится Норвегия, где благодаря обилию осадков и высоких гор в былые времена можно было любоваться красивыми и величественными картинами водопадов, а в настоящее время заставить служить их на пользу человечества. Для того, чтобы дать представление о количестве «белого угля», коим можно пользоваться на заводах в Норвегии приведем следующую таблицу:
|
Название водопада |
Количество воды в 1 секунду в кубометрах |
Высота падения, м |
Энергия, л. с. |
|
Svälgfos |
65 |
46,5 |
26500 |
|
Boolfos |
50 |
57,0 |
23500 |
|
Rjukan |
45 |
520,0 |
202500 |
|
Wamma |
220 |
20,8 |
36500 |
|
|
|
|
289000 |
Таким образом, Норвежская компания может располагать энергией в 300 000 лошадиных сил. Но по здешним сведениям число их должно быть повышено до 500 000 л. с. В настоящее время бывший водопад Rjukan исчез, ибо вода его направляется по 10 трубам, (см. рис. 5) свыше метра диаметром каждая, в силовой станции (расположенной на 300 метров ниже), и таким образом используется 120 000 л. с. Отработавшая вода из-под турбин образует еще второй водопад, падая метров на 100 почти вертикально, а потом еще большее падение имеет русло р. Маана, бурлящей наподобие Терека но каменистому ложу; чтобы использовать эту разницу уровней компания сверлит горизонтальный туннель в несколько верст в горе, чтобы провести воду к новой силовой станции Rjukan II, которая даст 107 000 лошадиных сил. На рис. 6 видна схема фабрикации азотной кислоты и ее солей на заводах Норвежской компании.
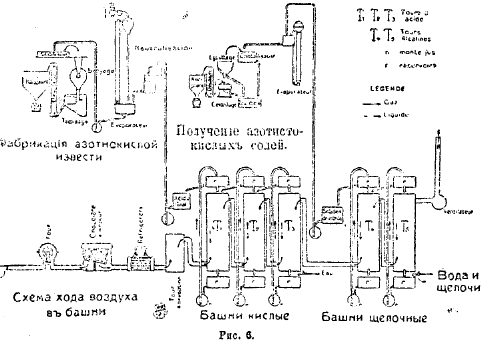
Рис. 6.
В печах Биркеланда и Эйде последней модели выход азотной кислоты на кВт-год равняется 600 килограммов. Поглощается водой 98%, следовательно, от 585 до 590 кило кислоты на кВт в год. Воздух вдувается в печи вентиляторами в количестве 25 000 литров в минуту на каждую печь (или 25 куб. метров). Выходящие из печей газы собираются в один канал, по которому они проходят к трубам парового котла, где охлаждаются до 200°. Пар, получающийся в котле, утилизируется для концентрации растворов азотнокислой извести.
Из под котла газы проходят через холодильные аппараты, в которых температура понижается до 50°—60°, и затем поступают в башни, в которых происходит окисление окиси азота в азотноватую окись (NО2):NO + O = NO2. Аппараты, необходимые для поглощения газов, поступающих из 3-х печей, состоят из двух групп, по 4 башни в каждой. Поперечное сечение башен равно 4 кв. метрам, а высота 10—20 метрам. Две первые башни каждой группы построены из гранитных плит, а две последние из песчаных. Все они наполнены кварцем или гранитными осколками, и газы проходят их одну за другой. Поглощение и концентрирование в башнях происходит следующим образом: в последней башне сверху струится вода, которая, поглотив азотную и азотистую кислоты, собирается в особый каменный собиратель откуда давлением воздуха поднимается в специальный ящик, находящийся на предпоследней башне. Слабая кислота, стекающая в этой башне, поднимается таким же образом на вторую башню и так же проходит ее, как в предыдущих. Выходящая отсюда уже довольно крепкая кислота пропускается, наконец, через первую башню, по выходе из коей достигает наивысшей концентрации, т. е. в ней содержится 50% HNO2. Газы, не поглотившиеся водой в башнях поглощения, проводятся в следующую башню, где циркулирует известковое молоко. Азотноватая окись поглощается известковым молоком, отчего получается смесь азотнокислого и азотистокислого кальция:
4NO2 + 2Ca(OH)2 = Ca(NO3)2 – Ca(NO2)2 + 2H2O.
В раствор солей пропускают горячий воздух, причем азотисто-кислый кальций переходит в азотнокислый. Наконец для того, чтобы удержать следы окислов азота, которые выходят из башни с известковым молоком, их проводят в меньших размеров башню, наполненную кусками негашеной взвести. Раствор азотнокислой взвести выпаривают на открытых сковородах до концентрации в 75—80% азотнокислого кальция (13,2—13,5% азота). Еще расплавленную жидкую массу разливают в железные бочки, где она застывает. Кроме того, в Нотоддене приготовляют основную азотнокислую известь с 10% азота, прибавляя к раствору соответствующее количество негашеной извести. Преимущество такой извести заключается в ее негигроскопичности. В последнее время газы из четвертой башни прямо пропускают в раствор едкого натра, причем от действия смеси окиси азота (NО) и азотноватой окиси получается азотисто-кислый натрий, или нитрит (NO + NO2 + 2NaHO = 2NaNO2 + H2O), имеющий большое применении в химической промышленности. Опыты Шлезинга показывают, что при пропускании газов, идущих из электрической, печи, через брикеты из гашенной извести, нагретые до 300°—350°, получается сухая кальциевая селитра (Ca(NO3)2) с небольшой примесью свободной извести, и азотистокислого кальция Ca(NO2)2.
Селитряный вопрос в России, в сожалению, находится только в стадии первоначальной разработки и, несмотря на неотложную важность разрешения этого вопроса для безопасности государства, он в течении более чем десяти лет обсуждался в комиссиях и хотя были получены удовлетворительные результаты с печью профессора А. И. Горбова и В. Ф. Миткевича, до сих пор (1917 г.) не построено завода, а производятся только предварительные работы.
Количество ввозимой в Россию селитры почти в 40 раз менее такового в Германию, как показывает следующая таблица:
1898 – 836442 пуд. или около 14000 тонн
1899 – 938402
1900 – 894146
1901 – 998755
1902 – 995866
1903 – 881007
1904 – 1038976
1905 – 1108169
1906 – 960876
1907 – 866801
1908 – 838228.
Около четверти вносимого количества, а именно – 150000-200000 пудов шло на потребности военного ведомства в мирное время. В военное время оно должно повыситься до 300000—350 000 пуд. (См. профессор А. Сапожников. Азотная кислота и селитра из воздуха. Стр. 50 — 52: Сиб. 1911), но так думали до 1914 г., когда еще не загорелся небывалый военный пожар, вышедший далеко за пределы Европы и потребовавший громадного количества взрывчатых веществ, следовательно, и азотной кислоты, необходимой для их приготовления. Селитра для России закупалась главным образом в Гамбурге, и еще в начале двадцатого столетия некоторыми более дальновидными людьми указывалось на необходимость освободиться от германской зависимости в этом отношении и озаботиться постройкой заводов для приготовления воздушной азотной кислоты, чтобы не очутиться в безвыходном положении в случае войны с Германией. В 1905 г. по приказанию генерал-инспектора по инженерной части была учреждена особая междуведомственная комиссия по добыванию азотной кислоты из воздуха. Председателем Комиссии состоял генерал-лейтенант Г. А. Забудский, делопроизводителем же А. И. Горбов. Вначале были проведены опыты с печью А. И. Горбова и В. Ф. Миткевнча в лабораториях Политехнического института профессором А. И. Горбовым и В. Ф. Миткевичем, а также были поставлены опыты превращения полученных окислов азота в азотную кислоту в химической лаборатории Михайловской артиллерийской академии профессором А. В. Сапожниковым.
После же того, как лабораторные опыты показали, что в печи А. И. Горбова и В. Ф. Миткевича выход окислов азота не уступает таковому в печах Биркеланда и Эйде и др. систем, опыты были произведены на Сестрорецком ружейном заводе в 1912—1913 году с печью А. И. Горбова и В. Ф. Миткевича мощностью до 200 киловатт. Они показали, что в названной печи при 10-часовой непрерывной работе при средней мощности дуги в 175 кВт выход (в среднем) азотной кислоты равняется 70,5 грамм на 1-й кВт-час, при содержании окиси азота (NO) в газах в 2,44% (объемных).
Таким образом, тип печи А. И. Горбова и В. Ф Миткевича является достаточно разработанным для того, чтобы перейти к заводской работе, так как выходы азотной кислоты в ней не хуже, а процент окиси азота выше тех, при которых работают в Норвегии и других странах.
Что касается вопроса об источниках энергии, необходимой для приготовления азотной кислоты, то, по мнению комиссии, наиболее подходящими для этой цели была бы водопады в Олонецкой губернии на реке Суне: Кивач, Пор-Порог и Гирвас.
Река Суна берет начало из озера Суно, близ границы Финляндии, проходит через озера Пор, Линдо, Суно (Суно тож) и впадает в Кондопожскую губу Онежского озера. Длина реки около 250 верст. Сплав леса россыпью производится по всей реке. Ниже Кивача судоходна. Ширина в верховьях от 5 до 10 саженей, в низшей до 40 саженей. Глубина от 1 до 6 саженей в порожистых от 1 до 1 ½ сажени. Водопад Кинач находится в 25 верстах от устья реки Суны, в 20 верстах от Кончезерского казенного завода. Водопад Пор-Порог на 61 версте, а водопад Гирнас на 62 версте от устья р. Суны.
Что касается мощностей этих водопадов, то можно иметь, при устройстве необходимых гидротехнических сооружений на Гирвасе и Пор-Пороге, 23 300 л. с. или 16 000 киловатт, а на Киваче 15 300 л. с. или 10 500 киловатт, т. е. энергией одного Кивача можно приготовлять в год от 250 000 до 300 000 пудов азотной кислоты.
Получение азотной кислоты окислением аммиака. Синтез окиси азота из азота и кислорода требует затраты энергии, и потому получение азотной кислоты синтетически может представлять выгоду только в тех случаях, когда имеются дешевые источники энергии. Окислы азота получаются при окислении аммиака, причем этот процесс сопровождается выделением тепла так, что сам может служить источником энергии. Процесс окисления аммиака известен давно; в учебниках химии описываются опыты, как горения аммиака, так и получения из него окислов азота: если смесь аммиака с воздухом пропустить через стеклянную трубку, в коей находится асбестовая пробка, покрытая мелкораздробленной платиной или же спираль из листовой платины, нагретые до 250-300°, то из трубки будут выходить окислы азота:
4NH3 + 5O2 = 4NO + 6H2O
Но получить подобным способом азотную кислоту в техническом масштабе удалось только в ХX в. Еще в 1839 г. Кульман предложил, пользуясь в качестве катализатора, платиной, получать азотную кислоту окислением аммиака. Дело не могло пойти вследствие того, что в то время аммиак был дорог. Но при развитии сухой перегонки каменного угля явился новый источник аммиака и его солей — промывные воды газовых заводов. С развитием интенсивного сельского хозяйства сернокислый аммоний нашел себе сбыт и потребление его, как и селитры, возрастало с каждым годом: количество сернокислого аммония, выпущенного на рынок во всех странах, было: в 1890 г. — 210 000 тонн, в 1900 — 493 000 т., а в 1910 — 1 112 000 т.
Благодаря способу Д-ра Монда количество аммиака, получаемого при коксовании каменного угля, можно увеличить в несколько раз. Вследствие этого в таких странах, как Россия, может получиться избыток аммиака, который не без выгоды можно будет превращать в азотную кислоту.
С другой стороны, в Германии уже с 1912 г. благодаря работам Габера и его сотрудников Баденской анилиновой и содовой фабрикой приготовляется синтетическим путем аммиак из водорода и азота. Кроме того, аммиак может получаться через разложение кальцийцианамида (см.), азотистого алюминия и др. металлов. Таким образом, источников получения аммиака множество, и, быть может, в недалеком будущем способ получения азотной кислоты из аммиака явится сильным конкурентом синтетического ее получения из азота воздуха.
В 1903 г. В. Оствальд задумал провести в техническом масштабе Кульмановский способ получения азотной кислоты, употребляя как катализатор платину. Прибор был устроен так, что смесь аммиака и воздуха перед тем, как вступить в соприкосновение с катализатором, обогревалась отходящими от него газами. Нужная для реакции температура катализатора поддерживалась теплотой реакции окисления. После реакции газы непосредственно вступали в поглотительные сосуды. Из испробованных Оствальдом катализаторов (окислов различных металлов, платинированного асбеста и т. п.) наилучшим оказалась белая блестящая платина: при ней 85% введенного аммиака окислялась в азотную (отчасти азотистую) кислоту и окись азота, 15% — совершенно терялось, превращаясь в азот и воду. Но предприятие В. Оствальда не получило широкого технического значения. Нужно отметить, что эти сведения имеются только до 1914 г. Что же касается до положения этого вопроса в настоящее время (1917 г.) в Германии, то, не имея возможности подучить точных сведений, мы можем только предположить, что ввиду потребности в азотной кислоте германские химики и техники, вероятно, с успехом разрешили задачу технического получения азотной кислоты из аммиака.
В России также этот вопрос может считаться решенным благодаря работам, произведенным в Центральной Научно-Технической лаборатории военного ведомства. Процесс окисления аммиака значительно улучшается при примешивании к воздуху и аммиаку перегретого пара. Воздух, предварительно нагретый в особой печи до температуры, которая определится природой употребленного катализатора (например, для платины 200°—250°), смешивается в особом смесителе с аммиаком и водяным паром, количество коего определяется дальнейшими условиями конденсации азотной кислоты в холодильниках. Добавочное, сверх указанного, количество водяного пара, вводится, когда контактное вещество начинает очень сильно накаливаться. Катализатор берется в виде одной или нескольких тонких платановых сеток или в виде спиральной колбаски из тонкой платиновой ленты. Можно употреблять также железную ветку, покрытую электролитической платиной. Что касается до аппаратов, служащих для поглощения образующихся окислов азота, то они могут быть построены подобно тому, как это было выше описано.
В России на юге количество аммиака и его солей (главным образом, сернокислого аммония) с развитием коксования каменного угля еще до войны увеличивалось с каждым годом, как показывает следующая таблица:
|
|
1911 г. |
1912 г. |
1913 г. |
|
Аммиачная вода (25%), пуд. |
188,6 |
308,8 |
734,9 |
|
Сернокислый аммоний, пуд. |
19886 |
135855 |
579650,0 |
|
Нашатырный спирт, пуд. |
8393 |
5120 |
6669,0 |
Сернокислый аммоний исключительно вывозился за границу; в Италию, Испанию и др. стр. Во время войны количество добываемого аммиака не уменьшилось, а значительно увеличилось, и поэтому представлялось выгодным превратить его в азотную кислоту иди аммиачную селитру (NH4NO2).
После того, как вышеуказанные лабораторные опыты оказались удачными, военным ведомством сперва был построен в Донецком бассейне небольшой опытный завод. Кроме того, было решено построить в Юзове завод с производительностью до 600 000 пуд. аммиачной селитры.
Литература. Donath und Frenzel, «Die Ausnutzung des atmosphärischen Stikstoffes» (Leipzig, 1907); Jurisch, «SaIpeter und sein Ersatz» (Leipzig, 1908); Guye, «La fixation industrielle de l’azote. Conference faite à la Société chimique de France, le 24 Mai 1909»; Grossman, «Die Stickstoffrage», 1911; Jean Escard, «La fabrication electrochimiqae de l’acide nitrique et des composés nitrés à l’aide des eléments de l’air» (Paris, 1909); Thomas H. Norton, «Utilization of atmospheric Nitrogen. Department of Commerce and labor. Special agents series. №52» (Washington, 1912); Сапожников, «Азотная кислота и селитра из воздуха» (Спб., 1911); А. Н. Саханов, «Получение азотной кислоты и ее солей из воздуха» (Москва, 1912); Ив. Каблуков, «Об использовании азота воздуха в целях питания» (Лекции, читанные на курсах для агрономов в 1912 г.); Кайзер, «Азот воздуха и его использование» (Изд. К. Л. Риккера, 1916).
Н. Каблуков.
| Номер тома | 38 |
| Номер (-а) страницы | 14 |